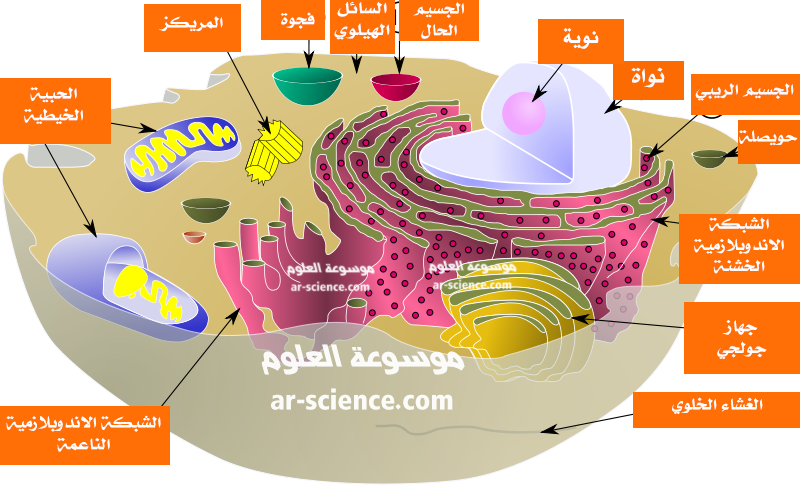
هو جزء من مادة الخلية الذي يقع بين الغشاء الخلوي والنواة . يتكون من حوالي 80% ما و 15% بروتينات ، كما انه يحتوي على دهون وسكريات واملاح معدنية بنسبة 5% ، وهو اقرب ما يكون الى نظام غروي يمتاز بلزوجته التي تختلف في المناطق المختلفة لنفس الخلية .
يعتبر السيتوبلازم الوسط الذي تحدث فيه تفاعلات كيميائية عديدة داخل تراكيب محاطة بأغشية يطلق عليها العضيات ، وكل منها يقوم بوظائف محددة . 1- الشبكة الاندوبلازمية :
تتكون الشبكة الاندوبلازمية من قنوات واكياس وحويصلات محاطة باغشية لها نفس تركيب الغشاء الخلوي والغشاء المكون للغلاف النووي حيث تشكل جهازا للتنقل الداخلي للخلية اضافة الى اعطاء الدعم للخلية .
وتنقسم الشبكة الاندوبلازمية الى نوعين :
أ – الشبكة الاندوبلازمية الخشنة :
وسميت خشنة لوجود حبيبات الرايبسومات على سطح غشائها ويكثر هذا النوع من الشبكة في الخلايا المتخصصة لصنع البروتين . وتتصل بالغلاف النووي .
ب- الشبكة الأندو بلازمية الملساء :
وسميت ملساء لان غشائها الخارجي يخلو من الرايبسومات . وتكثر في بعض الغدد مثل الغدة الكظرية والمبايض والخصي ولا تتصل بالغلاف النووي .
وظيفة الشبكة الاندو بلازمية :
1- تقوم بدور مهم في تنظيم تحويل الجليكوجين الى جلوكوز ؟
2- ازالة الاثر السمي لبعض العقاقير والسموم والادوية المخدرة وتختص بها بعض خلايا الكبد .
3- تساعد على عملية انقباض العضلات بسبب أختزانها لايونات الكالسيوم اللازمة لانقباض العضلة .
2- الرايبوسومات :
عضيات دقيقة توجد اما متصلة باغشية الشبكة الاندو بلازمية او حرة في السيتو بلازم ، وهي تتكون من البروتين والحمض النووي الرايبوزي ، ويكثر وجودها في خلابا الدم البيضاء .
الوظيفة : تعمل على انتاج وتكوين البروتينات .
3- جهاز جولجي
وهو عبارة عن تراكيب غشائية تحصر فيما بينها فراغات خلوية تشمل حزمة من اكياس مفلطحة مرتبة ترتيبا متوازنا ومن حويصلات غشائية ذات اغشية رقيقة وسميت نسبة الى مكتشفها عام 1873 م العالم جولجي .
4- الليسوسومات : (الاجسام المحللة )
الليسوسومات عبارة عن حويصلات غشائية بيضاوية الشكل غير منتظمة تنشأ من اجسام جولجي والشبكة الاندو بلازمية وتحتوي على انزيمات محللة تستخدمها الخلية لهضم الجزيئات الكبيرة كادهون والاحماض النووية والمركبات الكربوهدراتية . تكثر في خلايا الدم البيضاء والخلايا البلعمية لقدرتها على تحليل البروتيمات والاحماض النووية ، والسكريات بفصل الانزيمات الموجودة فيها .
وظيفة الليسوسومات :
1- التخلص من الاجسام الغريبة .
2- التخلص من الخلايا التالفة عن شيخوختها .
3- هضم الغذاء داخل الخلية كما في الكائنات وحيدة الخلية .
5- الميتوكندريا
وهو عبارة عن عضيات عصوية ، او كروية محاطة بغشاء مزدوج ينثني الى الداخل مكونا طيات او مخادع تدعى الاعراف . توجد بكثرة في انسجة العضلات والقلب والكبد وتحتوي على أنزيمات الاكسدة والتنفس وتنتشر في سيتوبلازم الخلايا كما توجد في الخلايا لعصبية والعضلية والافرازية .
وظيفة الميتوكندريا:
1- اكسدة المواد الغذائية وانتاج الطاقة (مركز لتحرير الطاقة) وتخزينها ولذا تجعى بيت الطاقة .
2- تساهم بعملية التنفس الخلوي لذا فالميتوكندريا تكثر في الخلايا التي لها علاقة بالطاقة والتنفس .
6- الفجوات :
تجاويف محاطة بأغشية فاذا كانت صغيرة الحجم أطلق عليها حويصلات . تنشا الفجوات من الشبكة الاندو بلازمية وجهاز جولجي . وتوجد فجوة او عدة فجوات منقبضة في السيتو بلازم (في الخلايا حقيقية النواة) وهناك انواع مختلفة من الفجوات ولكل منها اهميته ووظيفته .
واما في خلايا النبات : فتندمج الفجوات الصغيرة مكونة فجوة مركزية كبيرة تحتل معظم حيز الخلية ويحيط بها غشاء بلازمي يفصلها عن باقي تراكيب وعضيات السيتوبلازم وتسمى فجوة مركزية .
7- الجسم المركزي : السنتروسوم
جسم اسطواني يوجد في سيتوبلازم الخلايا الحيوانية (بالقرب من النواة) ، باستثناء الخلاي التي فقدت القدرة على الانقسام . كما يوجد في خلايا بعض الفطريات وقليل من خلايا بعض الطحالب مثل طحلب كلاميدوموناس ، ويوجد به جسيمان صغيران يعرف كل منهما بالجسيم المركزي .
التركيب : يتألف جدار الجسم المركزي من تسع مجموعات مرتبة في محيط واحد تضم كل مجموعة ثلاث انيبيات دقيقة متصلة معا .
الوظيفة : يقوم بدور اساسي في عملية الانقسام الخلوي ـ حيث يقوم بتكوين خلايا المغزل التي تظهر أثناء انقسام الخلية الحيوانية .
8- الهيكل الخلوي :
ما المقصود با الهيكل الخلوي؟ واين يوجد ؟ وما هي مكوناته ؟ وما هي وظائفه ؟
هو عبارة عن شبكة من الانيبيات والخيوط الدقيقة التي تقوم بتدعيم السيتوبلازم وتثبيت عضياته المختلفة حسب مواقعها المحددة (الحفاظ على شكل الخلية ودعمها) . وباستخدام المجهر الالكتروني امكن تمييز ثلاثة مكونات للهيكل الخلوي هي :
1- الانيبيبات الدقيقة :
يتكون من انابيب مجوفة من بروتين التيوبيولين والتي تلعب دورا في الحركة على اعلى مستوى الخلية كحركة الكروموسومات نتيجة تكوين خيوط المغزل .
2- الخيوط الوسطية :
وهي خيوط بروتينية ملتفة حول بعضها البعض كالحبل .
3- خيوط الاكتين الدقيقة :
تتكون من خيطين ملتفين حول بعضهما البعض من بروتين الاكتين وتلعب دورا مهما في انقباض العضلات والحركة الاميبية وانقسام الخلية
9-البلاستيدات :
ما هي البلاستديات ؟وما هي وظائفها ؟
البلاستيدات هي عبارة عن عضيات تتواجد في سيتوبلازم الخلية النباتية والطحالب فقط . وتصنف حسب وجود الصبغة الى الاتي :
1- بلاستيدات خضراء :
وهي تحتوي على مادة الكلوروفيل الخضراء وتوجد في الأوراق والاجزاء الخظراء وتقوم بعملية البناء الضوئي وخزن حبيبات النشاء.
2- بلاستيدات ملونة :
وتحتوي علة مواد صبغية ما عدا الكلورورفيل وتوجد في الأزهار والأثمار والسيقان وبعض الزهور
3- بلاستيدات عديمة اللون :
عضيات التخزين في النبات مثل درنات البطاطس حيث لا تحتوي على اصباغ .
تخزن النشا على هيئة حبيبات والبروتينات على شكل حبيبات .
تخزن الزيت والدهن على هيئة قطرات .







